StainedGlass

Identity heatmaps of genomic sequence
StainedGlass
![]()
![]()
![]()
![]()
This is a repository for making colorful identity heatmaps of genomic sequence.
Installation
To install you can follow the directions on the usage page or use the information below.
You will need a current version of snakemake to run this workflow. To get snakemake please follow the install instructions on their website, but in brief once conda and mamba are installed you can install snakemake with:
mamba create -n snakemake -c conda-forge -c bioconda 'snakemake>=8'
Afterwards you can activate the conda environment and download the repository. And all additional dependencies will be handled by snakemake.
conda activate snakemake
git clone https://github.com/mrvollger/StainedGlass.git
Running
Choose a sample identifier for your run e.g. chr8 and a fasta file on which you want to show the colorful alignments and the modify the config file config/config.yaml accordingly.
Once this is done and you have activated your conda env with snakemake you can run the pipeline like so:
snakemake --cores 24
Or do a dry run of the pipeline:
snakemake --cores 24 -n
All parameters are described in config/README.md and you can modify any of them
by modifying config/config.yaml. You can also change the configuration via the command line. For example, to change the sample identifier and fasta options do:
snakemake --cores 24 --config sample=test2 fasta=/some/fasta/path.fa
Please try the test case with the default configuration file before submitting issues.
If you are familiar with snakemake and want to trouble shoot yourself you can find the Snakefile in the directory workflow.
Output
The file results/{sample}.{\d+}.{\d+}.bed will contain all the alignments identified by the pipeline, and is the main input for figure generation. Under the same prefix there will also be a bam file that contains the unprocessed alignments. Note the bam will contain additional alignments not present in the bed file because redundant alignments with lower scores are removed before the figure generation.
Static dot-plots for moderate sized regions
To make pdfs and pngs for a particular set of regions just add make_figures to your command. This is generally appropriate for comparing up to ~5 regions totaling at most ~40 Mbp.
snakemake --cores 24 make_figures
This will make an output directory under results/{sample}.{\d+}.{\d+}_figures with a variety of dot plots in pdf and png format.
If you see tri.TRUE in the output pdf/png it means that the dot plot is rotated and cropped into a triangle. If you see onecolorscale.FALSE it means that between different facets in the same plot different color scales are being used.
Visualization of a large region or a whole genome
Making an interactive whole genome visualization requires the use of the program HiGlass and a web browser. However, this pipeline will make the necessary input files with the following command:
snakemake --cores 24 cooler
To view locally, use higlass-manage:
pip install higlass-manage
higlass-manage view results/small.5000.10000.strand.mcool
See the T2T CHM13 v1.0 StainedGlass for an example.
High resolution interactive visualization
To create a high-resolution interactive visualization where the coloring is proportionally to the number of reads mapped to each bin, use the following command:
snakemake --cores 24 cooler_density --config window=32 cooler_window=100
Arabidopsis: quick start, case example, and benchmark
To demonstrate a case example of using StainedGlass we applied the tool to a 132 Mbp chromosome level assembly of the Arabidopsis genome (DOI:10.1126/science.abi7489).
wget https://github.com/schatzlab/Col-CEN/raw/main/v1.2/Col-CEN_v1.2.fasta.gz \
&& gunzip Col-CEN_v1.2.fasta.gz \
&& samtools faidx Col-CEN_v1.2.fasta
Using 8 cores on a laptop with 32 GB of ram we ran StainedGlass using the following commands:
time snakemake --cores 8 --config sample=arabidopsis fasta=Col-CEN_v1.2.fasta
This command generated 41,036,963 self-self pairwise alignments within the assembly, 16,699,976 of which passed filters for downstream analysis.
Then to generate the cooler files that can be loaded in HiGlass we ran the following command with the already computed alignments:
time snakemake --cores 8 --config sample=arabidopsis fasta=Col-CEN_v1.2.fasta cooler
The results can be viewed at resgen.io/paper-data/Naish, and we include a static view of the centromeres here:
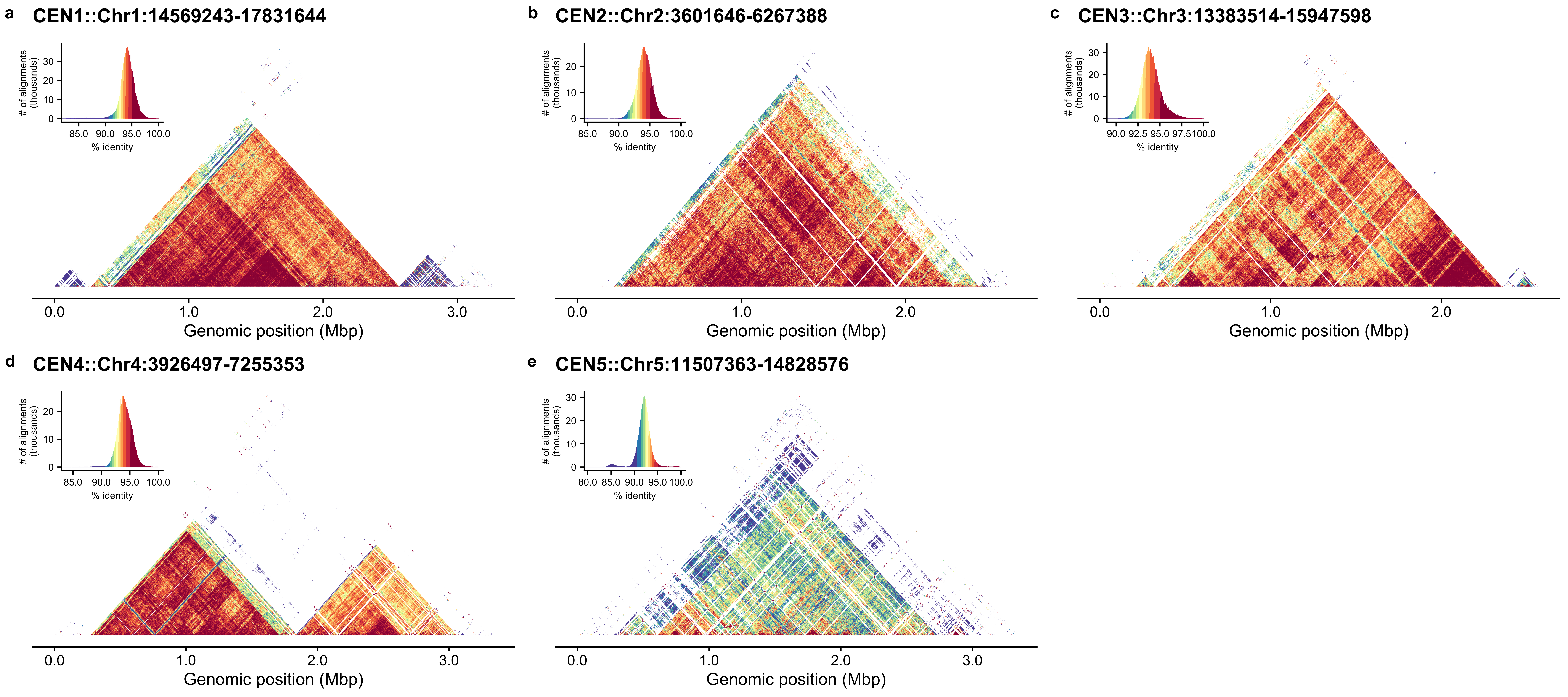
Arabidopsis runtime statistics:
| step | window (bp) | user (s) | system (s) | cpu (%) | wall (h:m:s) |
|---|---|---|---|---|---|
| alignment | 1,000 | 16,014.07 | 163.41 | 481 | 56:00.57 |
| cooler | 1,000 | 544.51 | 32.98 | 213 | 4:30.64 |
| static figures 1 | 1,000 | 2,635.30 | 188.07 | 58 | 1:20:14.59 |
A full report of all steps executed and the runtime of those steps is available in case-example-arabidopsis/report.html.
ARM Macs
Executing snakemake in the following way on ARM Macs may allow for bioconda to install the necessary dependencies:
CONDA_SUBDIR=osx-64 snakemake --cores all --use-conda
However, ARM Macs are not officially supported by StainedGlass at this time.
TODO
- Allow users to adjust the color pallet used in R
- Test short read aligners with smaller window sizes
- Make a more intelligent fragmentation method that won’t be affected by offsets in repeat motifs
- Consider alternative ways to score cells with multiple non-overlapping alignments
Cite
Mitchell R Vollger, Peter Kerpedjiev, Adam M Phillippy, Evan E Eichler, StainedGlass: Interactive visualization of massive tandem repeat structures with identity heatmaps, Bioinformatics, 2022; https://doi.org/10.1093/bioinformatics/btac018
-
Not recommended for whole genomes. ↩